Authors: Brianna Barrett, Ph.D., Associate Director, Technical Sales, Forge Biologics
Danielle Sexton, Ph.D., Scientist II, Forge Biologics
This article was first published in Endpoints News.
The field of gene therapy has been diligently moving forward over the past several decades to bring potentially life-saving treatments to patients with genetic diseases. In addition to two approved adeno-associated viral (AAV) gene therapies, there are more than 250 AAV gene therapies in various clinical trial stages.1 AAV vectors remain the most frequently used vector for delivering therapeutic transgenes to target tissues due to their demonstrated and lasting clinical efficacy and extensive safety track record. As AAV therapies advance through clinical trials and into commercialization, many biotech companies are turning to contract development and manufacturing organizations (CDMOs) to prepare their programs for late-stage clinical and commercial scale manufacturing. Given the scope and scale of the manufacturing needs that will accompany regulatory approvals for these assets, CDMOs continue to expand their capacity to meet the needs of increasing prevalent patient populations. However, despite rapid growth, projected gene therapy manufacturing demands still outpace the collective capacity of the CDMO industry.
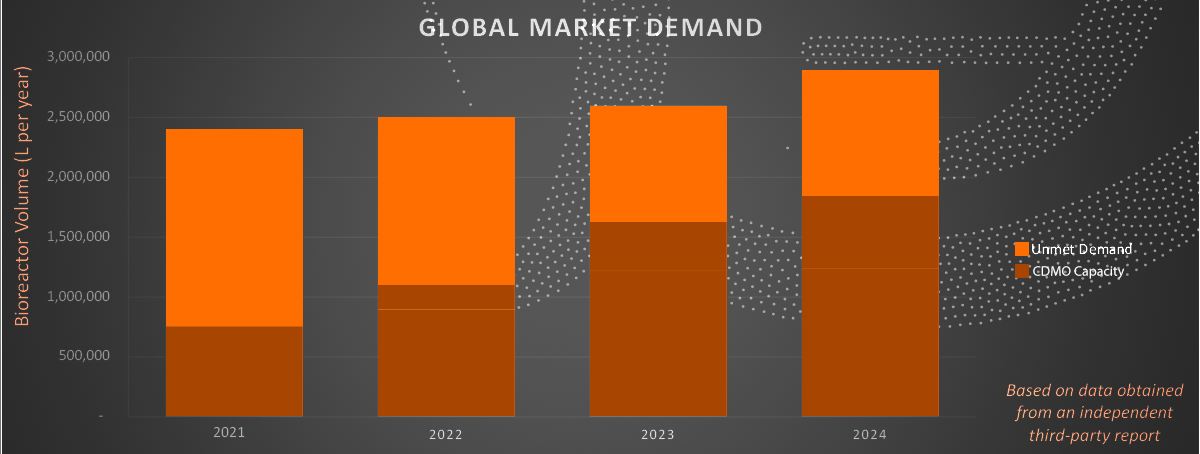 Global Market Demand. Data from an independent third-party report shows the persistent unmet gene therapy manufacturing demand, despite an increase in CDMO capacity over the next several years.
Global Market Demand. Data from an independent third-party report shows the persistent unmet gene therapy manufacturing demand, despite an increase in CDMO capacity over the next several years.
The Battle Between Time and Cost with a Custom vs. a Platform Process
Encouragement from regulatory agencies has prompted more CDMOs to adopt a platform process approach. As knowledge and experience from the past 40 years is applied, manufacturing strategies can be accelerated to increase access to life-saving gene therapies. However, terms such as ‘plug and play,’ or ‘turnkey,’ are over-simplified explanations of this approach. Instead, a platform approach with the flexibility to adapt to individual client needs can provide a head start on development that saves both time and money. On average, initiating development using a pre-existing manufacturing platform can result in a process reaching cGMP readiness approximately 30% to 60% faster than a process with different levels of customization. An additional advantage of a platform process is that the upfront investment of time and effort to characterize and continuously understand the process is the responsibility of the CDMO partner. This allows a client the flexibility to modify different variables, which may be recommended on a product-specific basis, and enables bespoke outputs. In this way, a platform process provides the best of both worlds: an approach that allows a quick path from the benchtop to the clinic but can address each product's unique considerations.
When is a Platform Process Recommended?
- When a platform process can be utilized to jump-start manufacturing with a known, easily produced product (such as a single-stranded, easily packaged AAV9), compared to a one-size-fits-all approach for potentially difficult products (such as a self-complementary, overstuffed AAV2)
- When a program requires small doses to achieve therapeutic outcomes
- When a program is targeting a disease with low incidence in the population
An example of a program with a low vector genome need is an AAV gene therapy for treating retinitis pigmentosa, an inherited retinal disease, which may require as little as 1e11 vg/eye. Yields from a small batch size (~50L working volume) using a platform process would provide enough material to treat hundreds of patients with a lower producing AAV2 vector, or thousands of patients with a higher producing AAV8 vector. In this instance, the immediate time savings and speed of therapeutic options for patients outweigh future cost efficiencies.
A different and more challenging example is a program requiring a large vector quantity, such as one targeting a patient population with muscular disease. This may require an upfront investment of time to increase transfection efficiency and productivity to maximize yield per batch. Performing this custom optimization may increase the feasibility of future product commercialization by reducing batch sizes or production frequency. For programs with high vector needs, this long-term vision and investment into process development should be seen as an asset being primed to become a successful commercial product. In this case, after upstream optimizations are performed, the program could still benefit from a platform downstream process. For example, each platform process unit step utilized for a product rather than a custom-developed step (such as lysis operation, clarification procedure, or empty/full separation technique) may save an average of 2-2.5 months.
How a Platform Process Accelerates Scale-up
Moving a program through research production, into clinical scale, and eventually commercial production requires a manufacturer to enable platform process synergies among process development, analytical development, and scale. Forge Biologics has demonstrated scalability from 1L to 1000L with multiple AAV serotypes through multiple iterations of regulatory interaction, facility developments, PD and AD evaluations and demonstrated cGMP productions. To date, Forge’s proprietary HEK 293 IgnitionTM cell line has scaled multiple natural and novel AAV serotypes successfully.
Two assets that reduce product-to-product variability in the performance of the platform process are Forge’s Ignition Cell Line™ and pEMBR™ Adenovirus Helper Plasmid. Reducing the variability of materials input into the system allows a smooth and predictable transition from 1L-5L-50-500L-1000L in single-use bioreactors.
- The Ignition Cell Line™ is a clonally derived HEK 293 suspension cell line that was screened for high AAV productivity and purity across numerous serotypes and selected for producing a high percentage of full AAV capsids.
- pEMBR™ is a proprietary Ad Helper plasmid with reduced adenoviral elements that has demonstrated equivalent AAV productivity, and an increased safety profile compared to commercially available Ad Helpers.
The Science of Scaling
Biologic manufacturing processes, specifically AAV, are divided into upstream and downstream events, each with diverse needs and goals. Upstream manufacturing focuses on scaling bioreactor parameters to allow cell growth and expansion, resulting in efficient and reliable AAV production, usually from a suspension culture. In contrast, downstream manufacturing focuses on purification and recovery of a highly concentrated, infectious AAV.
Bioreactor parameters such as power input/volume ratio (P / V), pH setpoint, temperature setpoint, and DO setpoint are held constant during upstream AAV manufacturing. Similarly, background air sparge, background air overlay, O2, and CO2 sparge rates are scaled proportionally to the working volume within the bioreactor. Bioreactor inputs such as plasmid molar ratio, viable cell density at time of transfection, transfection reagent to DNA ratio, DNA to viable cell ratio, and transfection cocktail working volume can all be pre-determined for a platform process. These inputs also scale proportionally with the working volume. In processes up to 500L, the necessary considerations for scaling bioreactor parameters are being increasingly demonstrated and understood throughout the industry. In processes from 1000L to 5000L, only a few groups, or in the case of 5000L volume, no groups to our knowledge, have yet attempted gene therapy manufacturing at such ambitious scales. At the 1000L+ scale, fluid dynamics and the coordination of moving, mixing, and storing large volumes of liquids become paramount.
During downstream AAV purification, careful assessment of yield recovery, purity and product quality are critical, as larger volumes of biomass need to be processed. If upstream parameters have been held consistent in a manner that produces equivalent biomass per volume, then scaling the clarification and affinity capture steps can be done by increasing filter surface area or resin volume to match the loading ratios (L / m2 or vg/mL) pre-determined by the platform process studies. However, as biomass increases, filter size and/or column size will need to increase to prevent filter or column fouling. A platform approach to affinity column chromatography does not necessarily impact scaling. Still, it affects the translation of the platform to multiple AAV serotypes, by using universal buffer matrices compatible with various column types. Empty/full capsid separation by CsCl density gradient ultracentrifugation is an efficient step for processes up to 500L, requiring a single day for processing. However, for processing 1000L+, there is value, or necessity, in reducing the volume of material after affinity elution to fit into 1-2 days of processing via an additional TFF concentration step. The final concentration UF/DF step is again scaled linearly by adjusting the filter surface area to the volume of material.
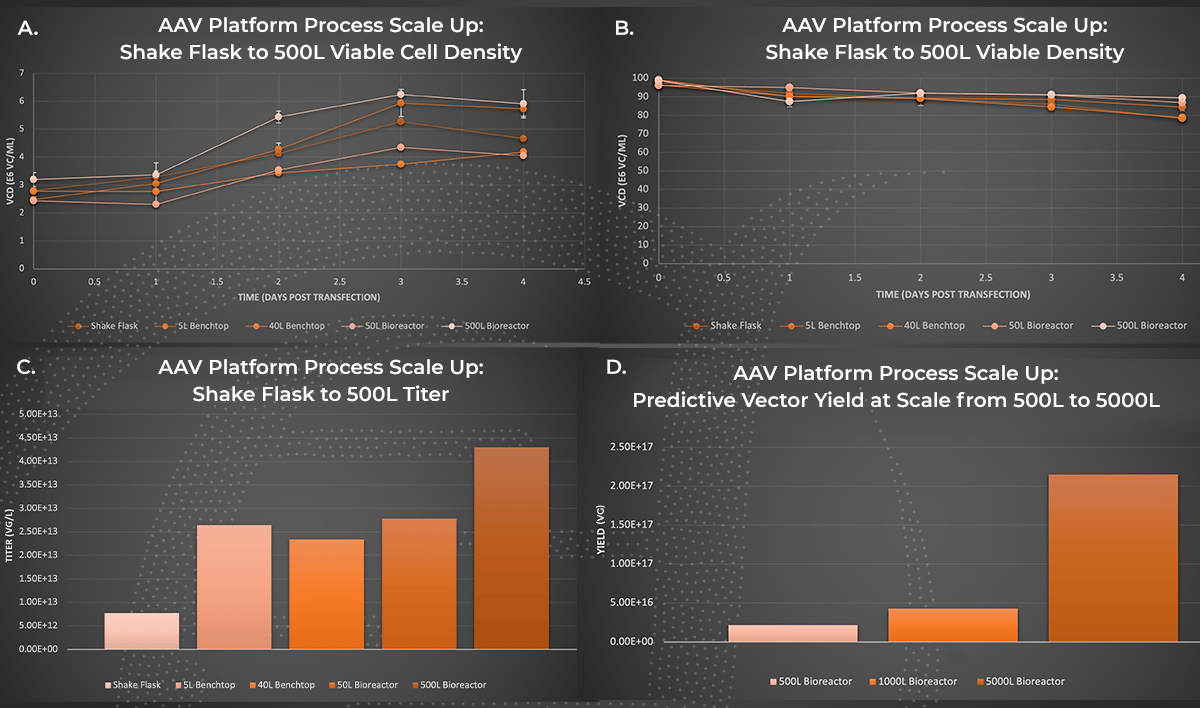
AAV Platform Process Scale Up. AAV manufacturing productions were performed in a 1L shake flask, or bioreactors with 5L to 500L working volume. Viable cell density (A.) and viability (B.) were measured on days one through four post-transfection with the Vi-Cell BLU cell counter. At day four post-transfection, cultures were harvested and purified through Forge’s platform downstream process and ddPCR titers of AAV in the clarified lysate material were obtained (C.). Extrapolated values representing clarified lysate titers for 1000L and 5000L production volumes (D.) were generated based on 1L to 500L volume AAV production trends.
Reaching the End Goal
Most gene therapy companies aim to provide patients with safe and effective treatment options as fast and cost-effectively as possible. Unfortunately, in the fast-paced biotechnology industry, the mission can often become buried in the day-to-day excitement of hitting a milestone or receiving a promising proof-of-concept result. While each step of the pre-clinical pathway is essential, a reminder of the end goal is always warranted. The accelerated timeline that accompanies utilization of an existing platform manufacturing approach, and an experienced CDMO, can be the difference that leads to timely treatment for many more patients with a long-term plan for manufacturing success.
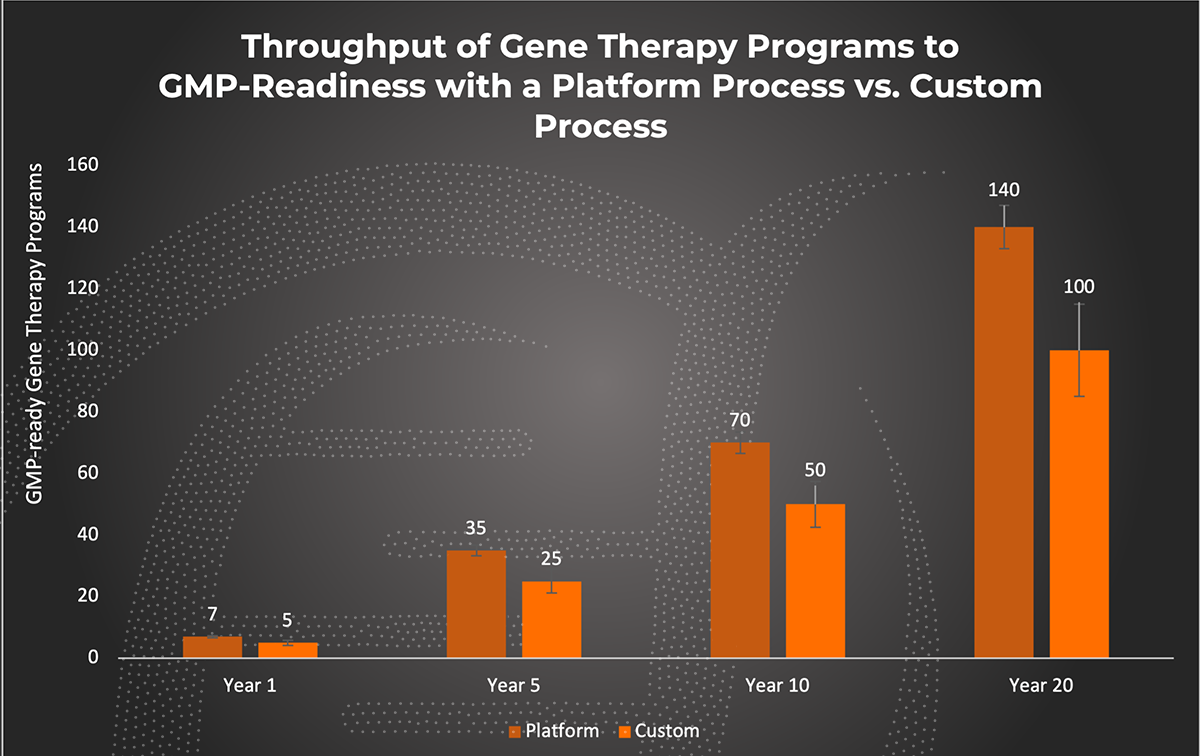
Throughput of Gene Therapy Programs. Estimations of gene therapy program throughput using a platform vs. custom process were generated based on average time spent in development, or pre-GMP activities. Time spent in pre-GMP activities for a platform process are estimated to range from 5-7 months compared to 8-12 months for a custom developed process.
- Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2021; 29: 464–488.
